A detailed introduction to rational design in synthetic biology
The concept of rational design
Rational design is a technique used to modify enzymes in synthetic biology, which is based on a deep understanding of the structure and function of the enzyme, by purposefully altering the amino acid sequence of the enzyme, thereby precisely regulating various properties of the enzyme, such as substrate specificity, catalytic activity, stability, selectivity, etc.
This design approach relies on knowledge and tools from biochemistry, structural biology, and computational biology to optimize enzymes or give them new functions based on pre-set goals.
Principles of rational design
1. Structural basis:
It is necessary to obtain the three-dimensional structure information of enzymes. This can be achieved by experimental means such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, or cryo-electron microscopy (cryo-EM) techniques.
Once the high-resolution structure of the enzyme is obtained, the spatial arrangement of its active sites, substrate binding sites, secondary structural elements, and various amino acid residues can be clearly seen.
This structural information is critical for identifying key catalytic residues and residues involved in substrate binding.
2. Computer Aided Design:
After the structural information is obtained, the molecular modeling and simulation software, such as PyMOL, Rosetta, Discovery Studio, etc., is used for computer-aided rational design.
Scientists can manipulate the structure of enzymes in a virtual environment to predict how changes in specific amino acid residues will affect the enzyme's function.
These software can simulate the substitution, insertion, or deletion of different amino acid residues, predict their effects on enzyme structure and function, and help researchers select the most promising mutation sites.
3. Functional mechanism understanding:
In addition to structural information, an understanding of the catalytic mechanism and function of enzymes is also key to rational design.
Understanding how enzymes interact with substrates, how they catalyze reactions, and which amino acid residues are involved in these processes can help researchers target mutations to change the enzyme's properties.
For example, if you want to change the substrate specificity of an enzyme, you might focus on amino acid residues in the substrate binding pocket;
If you want to improve the stability of an enzyme, you might want to consider key regions that affect protein folding and structural stability.
Steps to rational design
1. Structure analysis:
The three-dimensional structure of the target enzyme was analyzed by experimental technique. For some enzymes that are difficult to crystallize, other structural analysis methods, such as solid-state NMR or cryo-EM, can be tried to obtain structural information.
The obtained structural data were analyzed to identify key domains, active sites, substrate binding sites, and potential variable regions of the enzyme.
2. Mutation design:
Amino acid residues to be mutated are selected according to structural information and functional mechanism.
It can be based on existing knowledge of structure-function relationships, or reference to structural information of other enzymes with similar functions.
Molecular modeling software is used to design different mutation schemes and predict the effects of these mutations on enzyme structure and function.
Single point mutations, multipoint mutations, or more complex structural rearrangements can be considered.
3. Expression and screening:
The engineered mutant gene is cloned into an appropriate expression vector, and the mutant enzyme is expressed in a suitable host cell, such as E. coli, yeast, or mammalian cells.
Establish a screening system to detect whether the mutant enzyme achieves the expected performance improvement.
Screening methods may include enzyme activity assays, substrate specific tests, stability assessments (e.g. activity tests at different temperatures, pH or organic solvents), etc.
Here are some commonly used tools and techniques for rational design in synthetic biology:
Structural analysis technique
1. X-ray Crystallography:
Principle: The protein crystal is placed in an X-ray beam, and the X-rays are scattered by the atoms in the protein. By measuring the intensity and direction of the scattered X-rays, the atomic structure of the protein can be resolved through complex mathematical calculations.
Advantages: Can provide high resolution (usually up to 1.5 A or higher) protein structure information, clearly show the location of atoms and chemical bonds, is a classic technique for studying protein structure.
Limitations: Proteins are required to be able to form high-quality crystals, and applications may be limited for some proteins that are difficult to crystallize (such as membrane proteins) or proteins with dynamic structures.
2. Nuclear Magnetic Resonance (NMR) spectroscopy:
Principle: By using the magnetic resonance phenomenon of atomic nuclei, the three-dimensional structure of proteins can be inferred by measuring the chemical shift and coupling constant of atomic nuclei (such as hydrogen, carbon, nitrogen, etc.) in proteins.
Suitable for relatively small proteins (usually less than 30 kDa), it is very effective for studying protein structure in solution.
Advantages: Can study the dynamic structure of proteins in solution, without crystallization, can provide information about the dynamic characteristics of proteins, closer to its physiological state.
Limitations: The study of proteins with larger molecular weights is difficult, the experimental operation is complex, and the sample purity and concentration requirements are high.
3. Cryo-Electron Microscopy (cryo-EM) :
Principle: A protein sample is quickly frozen, the sample is imaged at low temperatures using an electron beam, and the three-dimensional structure is reconstructed through computer processing of a large number of two-dimensional projected images.
Advantages: It can handle large protein complexes, and has unique advantages for proteins that are difficult to crystallize and proteins with dynamic structures, and the resolution has been continuously improved in recent years, and even can reach a resolution close to the atomic level.
Limitations: Instruments are expensive, image processing and structural reconstruction require powerful computing resources and specialized software.
Computer aided Design software - molecular modeling software
1、PyMOL:
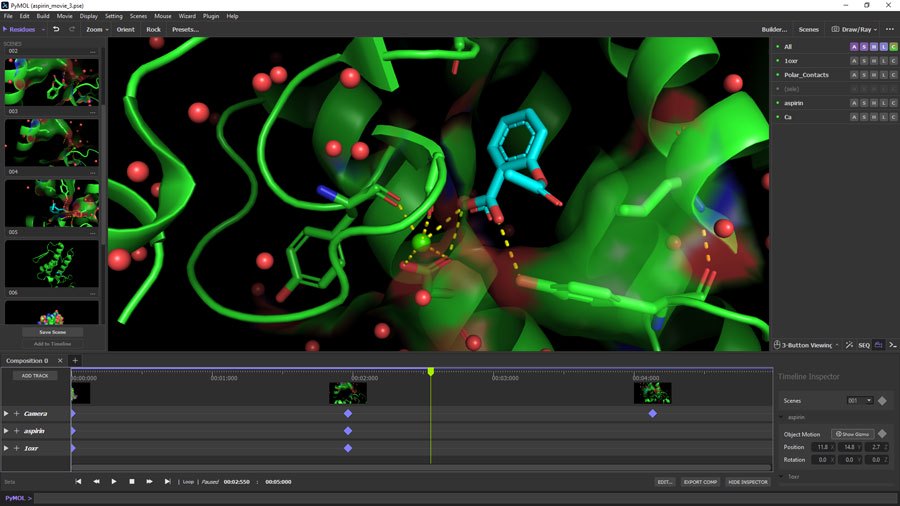
Function: It integrates the functions of molecular modeling, simulation and drug design to provide an integrated working platform.
It can visualize and edit protein structure, perform molecular dynamics simulation, virtual screening, drug design, protein-ligand interaction analysis, etc.
Features: Fully functional, with a variety of algorithms and tools, suitable for research from structural analysis to drug design and other aspects of research, suitable for drug development teams and synthetic biology involving small molecule and protein interactions.
2、Rosetta:
Function: It is a powerful protein structure prediction and design software package that can not only predict the structure of proteins, but also carry out protein design and optimization.
It can carry out de novo design, protein-protein docking, protein-small molecule docking, enzyme active site design and so on.
Features: Through a large number of algorithms and calculations, it can carry out complex operations and predictions on protein structure, such as folding proteins from the ground up to predict the optimal structure under different amino acid sequences;
The active site of the enzyme was designed to predict the best amino acid conformation for substrate binding and catalysis.
3、Discovery Studio:
Function: A widely used molecular visualization software that can load protein structure files (such as PDB files), display, analyze and edit the structure of proteins.
It can display atoms, bonds, secondary structures, surfaces, etc., which is convenient for users to observe and analyze protein structure.
At the same time, simple mutation simulation can be carried out to predict the change of protein structure after mutation.
Features: simple and intuitive operation, friendly interface, suitable for beginners and structural biologists to quickly observe and preliminary analysis of the structure, but the function is relatively basic.
Molecular Dynamics (MD) simulation
Principle: Through the principle of Newton mechanics, the physical movement of atoms and molecules is simulated on the computer, and the position and speed of each atom in the molecular system are calculated according to the laws of physics, so as to study the dynamic behavior of the molecular system.
Advantages: It can study the dynamic process of protein, such as protein folding, conformational change, binding process with substrate, stability in different environments, etc., providing dynamic information that cannot be obtained by structural analysis technology.
Limitations: It is very computationally intensive, requires high performance computing resources, and the time scale of simulation is usually short, making it difficult to simulate long-term biological processes.
Quantum chemical computation
Principle: Based on the principle of quantum mechanics, the precise calculation of the electronic structure of molecules can be used to study the chemical mechanism of enzyme-catalyzed reactions, including the electron distribution of the substrate, the nature of the transition state, and the reaction energy barrier.
Advantages: The catalytic mechanism of enzymes can be understood from the level of atoms and electrons, providing a theoretical basis for the design of the active site of enzymes, and accurately calculating the energy change of the reaction.
Limitations: Very computationally intensive, only applicable to small systems (such as active site regions), extremely high computational resource requirements, requiring professional quantum chemistry software and knowledge.
Bioinformatics tool
BLAST (Basic Local Alignment Search Tool) :
Function: Used for similarity search of protein or nucleic acid sequences to help find homologous sequences.
By sequence alignment, homologous proteins with known functions can be found, which can provide reference for structural and functional analysis of target proteins.
Features: It is the most commonly used sequence comparison tool, simple and easy to use, and can be used online through NCBI and other websites to provide sequence information support for rational protein design.
UniProt:
Function: A comprehensive protein database containing a large amount of protein sequence and structure information, as well as annotation information (function, subcellular localization, modification, etc.).
Features: It provides abundant protein information resources for rational design, and can query the known information of target proteins, such as known functional domain, structural domain, known mutation information, etc.
These tools and techniques are often used collaboratively in rational design, from structural analysis to computer-aided design, dynamic simulation and bioinformatics analysis, providing a full range of support for rational design of enzymes from structure to function, helping scientists to accurately modify enzyme performance and promote the development of synthetic biology.
In practical applications, researchers will choose the right combination of tools and technologies according to the specific research objectives and conditions to achieve accurate transformation and optimization of enzymes.
For example, when studying a new enzyme, BLAST and UniProt are first used to find homologous sequences and known information, then the structure is resolved using X-ray crystallography or cryo-EM, then the design is performed using Rosetta, and finally the effect of the design is evaluated by molecular dynamics simulations.
Rational design application team success stories
Case 1: Transformation of industrial enzymes
Team: Enzyme engineering team of a large biotechnology company.
Objective: To improve the stability and activity of an industrial lipase in an organic solvent for lipid hydrolysis and esterification in biodiesel production.
Procedure:
1. The structure of the lipase was analyzed by X-ray crystallography, and the active sites and areas that may affect stability were identified, especially the surface areas exposed to solvents.
2. Based on structural analysis, a series of mutations were designed using molecular modeling software, focusing on hydrophobic amino acid residues on the surface and some residues involved in the hydrogen bond network of the enzyme structure stabilization.
For example, some hydrophilic residues are replaced with hydrophobic residues to enhance the compatibility of enzymes with organic solvents; At the same time, the hydrogen bond networks of key domains were adjusted to improve the overall stability.
3. The designed mutant gene was expressed in E. coli, and its activity and stability were tested in a reaction system containing different organic solvents.
Result:
Several mutants with significant improvement were successfully screened, and one double mutant showed 3 times higher activity than wild type enzyme in high concentration organic solvent, and its stability was improved by about 2 times, which greatly improved the efficiency and economy of biodiesel production.
Case 2: Modification of drug metabolizing enzymes
Team: A biomedical research group at a university.
The goal: To engineer a cytochrome P450 drug metabolism enzyme to metabolize a newly developed drug precursor, converting it into an active drug form while avoiding interference with the body's normal metabolic pathways.
Procedure:
1. Based on homology modeling and known P450 enzyme structure information, a three-dimensional structure model of the drug metabolizing enzyme was constructed.
2. The substrate-binding pocket was analyzed in detail and a series of mutations targeting amino acid residues of the substrate-binding pocket were designed to expand the size of the pocket and change its chemical properties to make it more suitable for the binding and catalytic conversion of new drug precursors.
3. The mutant enzyme is expressed in mammalian cells to evaluate its ability to metabolize new drug precursors and its selectivity to normal substrates through drug metabolism experiments.
Result:
After several rounds of rational design and screening, a mutant was obtained, which could effectively transform drug precursors into active drugs without significant changes in the activity of normal metabolic substrates, providing important support for the development of this new drug.
Case 3: Enzyme design in biosensors
Team: An interdisciplinary synthetic biology research team.
Objective: To design an enzyme biosensor that can be used to detect environmental contaminants, a REDOX enzyme needs to be modified to have high specificity and sensitivity to low concentrations of contaminants.
Procedure:
The crystal structure of the oxidoreductase was analyzed, and its binding process to pollutants and natural substrates was studied by molecular dynamics simulation.
Several key amino acid residues were identified and their charge and spatial properties were changed by rational design to enhance their specific binding and catalytic capacity to contaminants.
The modified enzyme was combined with an electrochemical sensor to test its response under different pollutant concentrations.
Result:
The resulting mutant enzyme biosensor can detect extremely low concentrations of pollutants, the detection limit is an order of magnitude lower than that of the original enzyme, and the cross-reaction to other similar substances is significantly reduced, providing a sensitive and specific biosensor for environmental monitoring.
These success stories show that rational design has great potential for enzyme modification in synthetic biology, which can precisely tune enzyme properties to bring innovative solutions to multiple fields such as industry, biomedicine and environmental monitoring.
But at the same time, it should be noted that rational design depends on high-quality structural and functional information, as well as accurate computational simulation, and in practical applications may require multiple rounds of design and optimization to achieve the ideal effect.