Glutathione promotes T cell metabolism, which leads to inflammation

abstract
Activated T cells produce reactive oxygen species (ROS), which trigger the antioxidant glutathione (GSH) response, which is essential for buffering elevated ROS and preventing cell damage.
We report that GSH is essential for T cell effector function by regulating metabolic activity. Conditional gene knockout of the catalytic subunit of glutamate-cysteine ligase (Gclc) blocks GSH production in mouse T cells.
T cells lacking Gclc are initially activated normally, but are unable to meet their increased energy and biosynthesis needs.
Glutathione deficiency impimpes the activation of mammalian rapamycin target-1 (mTOR) and the expression of NFAT and Myc transcription factors, thereby impacting energy utilization and MYC-dependent metabolic reprogramming, in which Myc metabolic reprogramming switches activated T cells to glycolysis and glutaminolysis.
In vivo, T cell-specific knockout of Gclc protects against autoimmune disease, but blocks antiviral defenses. Thus, the antioxidant glutathione pathway plays an unexpected role in metabolic integration and reprogramming during inflammatory T cell responses.
result
1. Glutathione is not necessary for early T cell activation, but can promote T cell growth
Since proliferating T cells accumulate ROS, the authors speculate that the glutathione pathway in these cells may be up-regulated. naive wild-type (WT) mouse T cells were activated with aCD3+aCD28 in vitro, and Gclc and Gclm mRNA were measured by quantitative RT-PCR.
After TCR triggering, Gclc mRNA was upregulated, but Gclm mRNA was not. The addition of butylthionine sulfoxide (BSO), a specific inhibitor of GCL activity, inhibited upregulation of Gclc, suggesting that the glutathione pathway was activated by TCR triggering.
Produced Cd4creGclcfl/fl mice in which Gclc was specifically deleted in T cells, and juvenile CD4+ and CD8+ T cells isolated from spleen and lymph nodes (LN) of GCLC-specific knockout mice had very low glutathione content. But 24 hours after activation with aCD3+aCD28 in vitro or when mice were injected with anti-CD3 or Staphylococcal enterotoxin B (SEB) to activate T cells in vivo, glutathione in these cells increased dramatically.
Cd4creGclcfl/fl mice showed normal thymus cell development, but decreased peripheral CD8+ and CD4+ T cells.
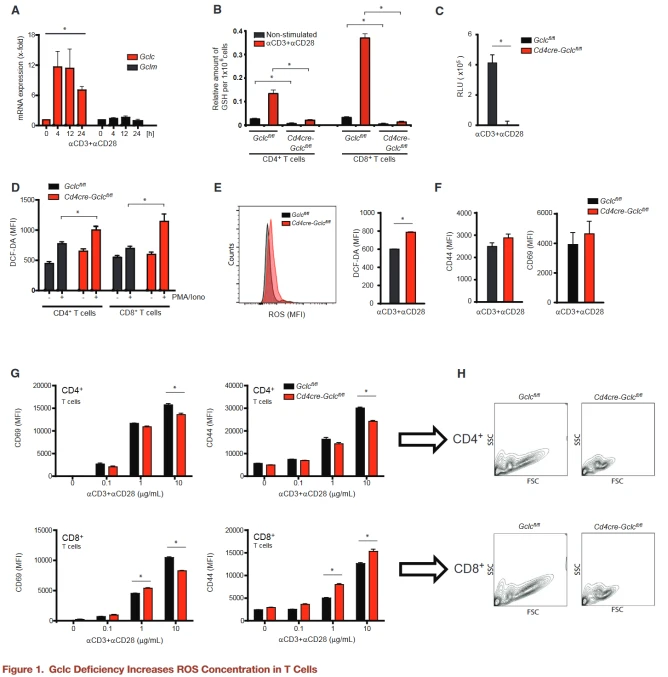
Gclc is important for T cell homeostasis and glutathione synthesis. The authors expected Cd4creGclcfl/fl T cells to accumulate high concentrations of ROS, but flow cytometry found only a slight increase in ROS concentrations in GCLC-deficient T cells activated in vitro or in vivo.
Expressions of CD69 and CD44 on T cells of both genotypes were comparable in vivo and in vitro, indicating that Gclc is not important for the initial phase of T cell activation.
Gclc deficiency reduces the ability of activated T cells to become large cells, both in vitro and in vivo, and increases cell mortality after 48 hours of stimulation.
Glutathione is not essential for initial T cell activation, but controls T cell homeostasis in vivo and promotes activation dependent growth.
T cell derived Gclc is required for mTOR activation
When T cells are activated, the cells exhibit an increase in cell volume, which is controlled by the mammalian target protein Complex-1 (mTORC1) of rapamycin.
The authors therefore investigated whether Gclc deficiency reduced mTOR activity in CD4+ and CD8+ T cells stimulated with aCD3+aCD28 in vitro.
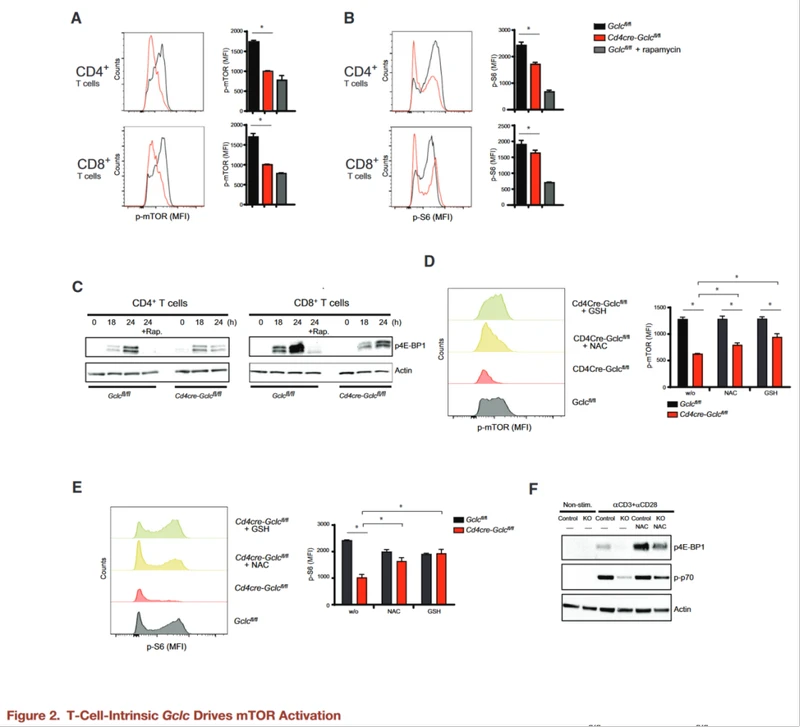
Flow cytometry and WB confirmed that Gclc deficiency blocks phosphorylation of mTOR S2448 and mTORC1 targets S6 and 4E-BP1, an effect consistent with that of the mTOR inhibitor rapamycin.
Cells are known to control mTOR activity by inhibiting tuberous sclerosis Complex 2 (TSC2) phosphorylation by AKT or GSK3β, but the authors found no difference in AKT or GSK3β activation between control and mutant T cells.
To test whether elevated ROS reduced mTOR activity in Cd4creGclcfl/fl T cells, T cells of both genotypes were stimulated with aCD3+aCD28 and treated with GSH or NAC, another antioxidant.
Elevated ROS concentrations in activated GCLC-deficient T cells were reduced to near WT concentrations by NAC or GSH, and mTOR activation and phosphorylation of S6, p70, and 4E-BP1 were partially restored. This suggests that these signaling changes in GCLC-deficient T cells are primarily due to the lack of ROS buffering.
Gclc supports glutamine metabolism
mTOR responds to nutrient deficiency by stimulating the synthesis of proteins, nucleotides, and lipids, and the authors speculate that the lack of Gclc in T cells may inhibit mTOR activation and glutamine utilization.
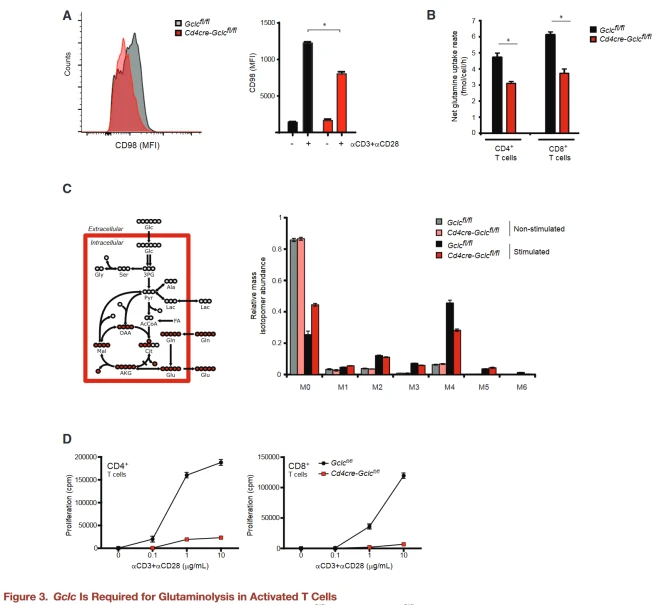
CD98 is a heterodimeric amino acid transporter that is induced in activated T cells and is essential for glutamine absorption. The authors found that activated Gclc deficient T cells expressed less surface CD98 than controls.
By calculating the molar difference between glutamine absorption and glutamate secretion to calculate the amount of glutamine entering the TCA cycle, the net influx of glutamine-derived carbon in GCLC-deficient T cells was reduced compared to the control group.
Next, mutant and control T cells were activated with aCD3+aCD28 in the presence of U-13C-glutamine, and their incorporation into downstream metabolites was determined by analyzing the mass isotope distribution (MID) of TCA cycle intermediates under metabolic and isotopic homeostatic conditions.
Although the relative flux of glutamine into TCA intermediates in activated T cells of both genotypes was higher than the homeostasis value, this increase was less in GCLC-deficient T cells.
Because glutamine is essential for T cell proliferation, the authors further compared the proliferation capacity of activated Gclc knockout and control T cells.
Gclc knockout T cells were undergrown after aCD3+aCD28 stimulation, and this reduced growth was not due to increased cell death.
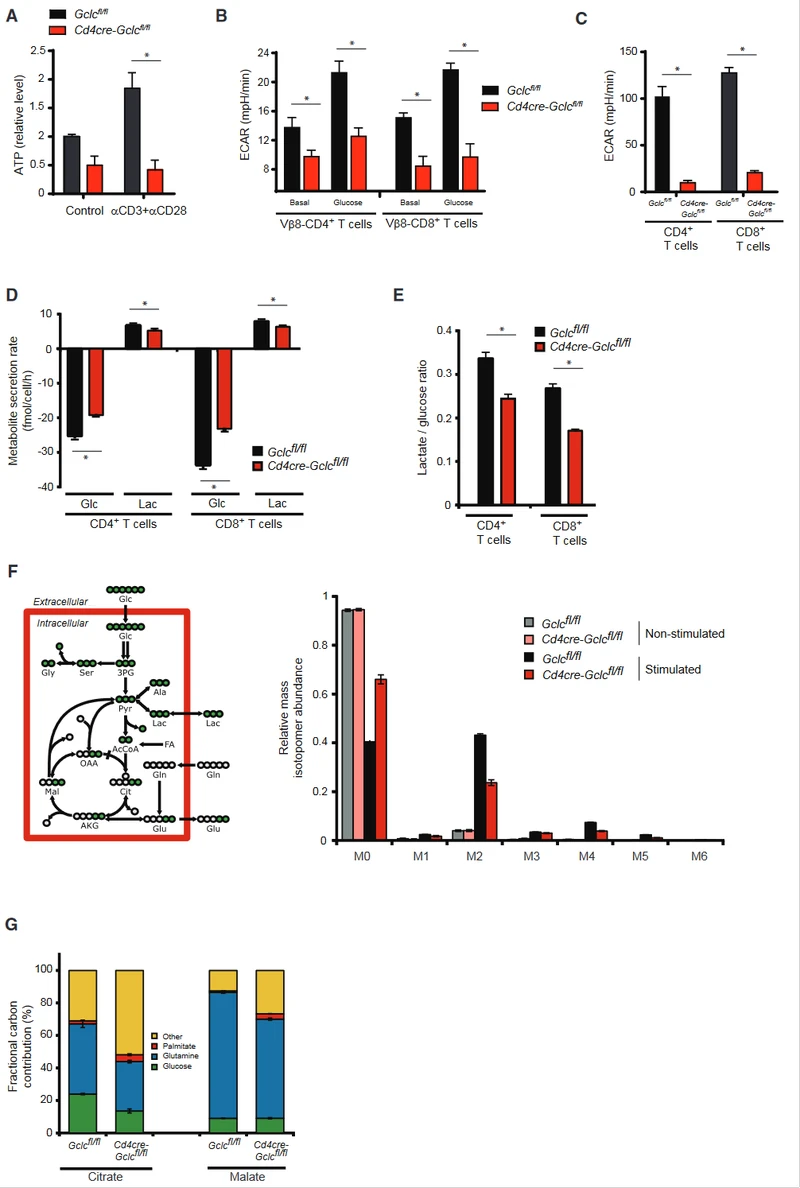
Gclc regulates metabolic reprogramming
Proliferating T cells require fatty acids to synthesize lipids and membranes. These components are provided by increased glycolysis and glutamine breakdown, which also fill the cellular energy bank with ATP.
To investigate whether Gclc loss affected the energy pool, the authors measured ATP in activated control and Gclc deletion mutant T cells.
Compared with controls, intracellular ATP was reduced in the resting state Gclc deletion mutant T cells, and ATP produced by Gclc deletion T cells did not increase after activation.
To determine whether Gclc regulates aerobic glycolysis, measured by extracellular acidification rate (ECAR), Gclc ablation reduced glycolysis in in vivo or in vitro activated mutant T cells.
Glutathione plays an unexpected role in regulating glucose metabolism. Glucose consumption and lactate secretion were then measured in activated Gclc deficient and control T cells over a 24-hour period.
Gclc-deficient T cells consumed less glucose and produced less lactic acid than controls, and the ratio of lactate secreted to glucose consumed (a measure of glycolytic ATP production) was reduced.
Gclc defects reduced the flux of glycolyso-derived pyruvate through lactate dehydrogenase (LDH) during the 24-hour activation period, indicating an altered flux of glucose into the TCA cycle through pyruvate dehydrogenase (PDH).
When resting control and Gclc deficient cells were incubated with U-13C-glucose, an equivalent fraction of the M2 isotope of citric acid was observed, which represents the relative flux of glucose-derived carbon through PDH.
After activation, the flux of PDH increased substantially in T cells of both genotypes, but decreased citric acid isotope M2 was found in GCLC-deficient T cells, indicating decreased PDH activity.
Compared to M2 citric acid, there was no reduction in the TCA-cyclically derived M1 citric acid isotope in the absence of Gclc. M1 citric acid is produced by the subsequent cycle of M2 citric acid, and the M1:M2 citric acid ratio indicates TCA cycle activity.
Despite GSH defects in mutated T cells, overall TCA cycling activity increased, which was consistent with glutamine tracking experiments.
Although the relative contributions of glucose and carbon glutamine to the TCA cycle decreased when GSH was deficient, the overall activity of the TCA cycle increased.
This result can only be explained by the continuous replenishment of the mitochondrial acetyl-CoA reservoir by another carbon source.
To test whether this source was an increase in fatty acid oxidation, the authors incubated Gclc deficient cells and control CD4+ T cells with U-13C-palmitic acid, which was transported to the mitochondria and oxidized to produce acetyl-CoA.
In fact, fatty acid beta oxidation increased in GCLC-deficient T cells compared to controls.
Carbon sources other than glutamine, glucose, and palmitic acid still contribute significantly to TCA cycle intermediates, and this contribution is greater in GCLC-deficient T cells than in controls.
These sources may be fatty acids (including palmitic acid) from FBS in the medium or fatty acids produced by catabolism of branchchain amino acids.
During metabolic reprogramming of activated T cells, mitochondrial oxidative phosphorylation increased, but consistent with a decrease in ATP, activated GCLC-deficient T cells had a lower oxygen consumption rate (OCR) than controls.
The unknown additional carbon source cannot make up for the energy gap in T cells that Gclc lacks. Therefore, glutathione is essential for metabolic reprogramming during T cell activation.
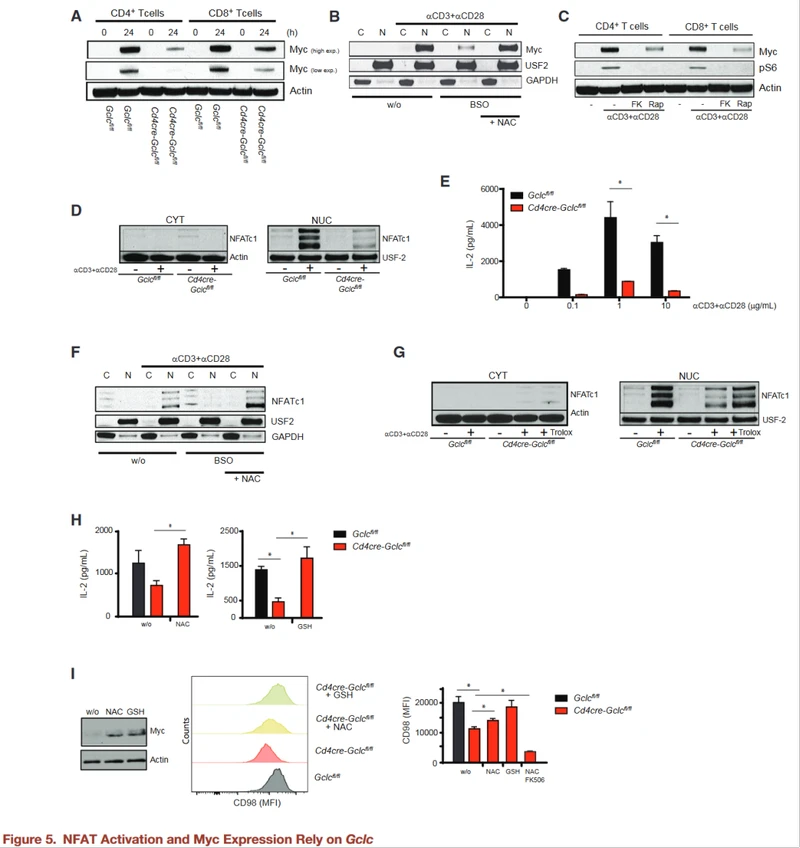
Loss of Gclc impairs TCR-induced Myc expression and NFAT activation
The transcription factor Myc is involved in activation induced glycolysis and glutaminolysis in T cells. Gclc deficiency reduces TCR-driven Myc upregulation.
To simulate Gclc deficiency in WT T cells treated with the inhibitor BSO, BSO-treated activated WT T cells showed reduced GSH after 24 hours and accumulated higher ROS than untreated WT T cells.
Bso-treated WT T cells also showed a reduction in Myc, while NAC co-incubation and ROS clearance restored Myc. Myc controls the variable splice-mediated transition from pyruvate kinase PKM1 to PKM2 expression and promotes glycolytic lactic acid production.
The authors found that PKM2 protein was down-regulated in Gclc knockout T cells, which was consistent with altered PDH glucose flux and reduced lactic acid production in these cells.
Next attempts were made to determine which pathway mediates GSH-dependent Myc expression in T cells. NFAT is known to promote Myc production in cancer cells, but the situation in T cells is unclear. NFAT also drives mTOR expression, while mTOR stabilizes Myc translation.
The authors stimulated WT T cells with aCD3+aCD28 plus the calcineurin inhibitor FK506 (which weakens NFAT activation) or rapamycin. WB showed that inhibition of NFAT or mTOR decreased Myc protein, while S6 phosphorylation was inhibited by FK506.
NFAT controls mTOR activity and Myc production in T cells. In addition, nuclear accumulation of NFAT was significantly reduced in activated GCLC-deficient T cells compared to controls, despite similar levels of Ca2+ mobilization.
Gclc-deficient T cells secrete less IL-2 (NFAT target and T cell mitogen). However, adding IL-2 to GCLC-deficient T cell cultures did not restore normal proliferation, suggesting that lack of IL-2 is not the primary factor preventing the expansion of mutant T cells. BSO reduced nuclear NFAT in activated WT T cells, but not cytoplasmic NFAT.
Calcasterin typically dephosphorylates NFAT to allow it to enter the nucleus (Rusnak and Mertz, 2000). The active site of calcineurin contains iron and zinc, which are sensitive to high ROS (Namgaladze et al., 2002).
The authors found that WT T cells exposed to BSO treated with NAC to induce ROS clearance were susceptible to NFAT nuclear translocations.
ROS buffering in GCLC-deficient T cells restores NFAT expression and nuclear translocation, as well as activation-induced IL-2 secretion.
Adding NAC or GSH to GCLC-deficient T cells increased the expression of Myc and its target CD98 in a manner that was sensitive to FK506 inhibition.
NFAT activation, Myc expression, and CD98 production in activated T cells require an antioxidant environment.
The above data suggest that ROS buffering of GCLC-derived GSH enables NFAT and mTOR to control the metabolic regulator Myc.
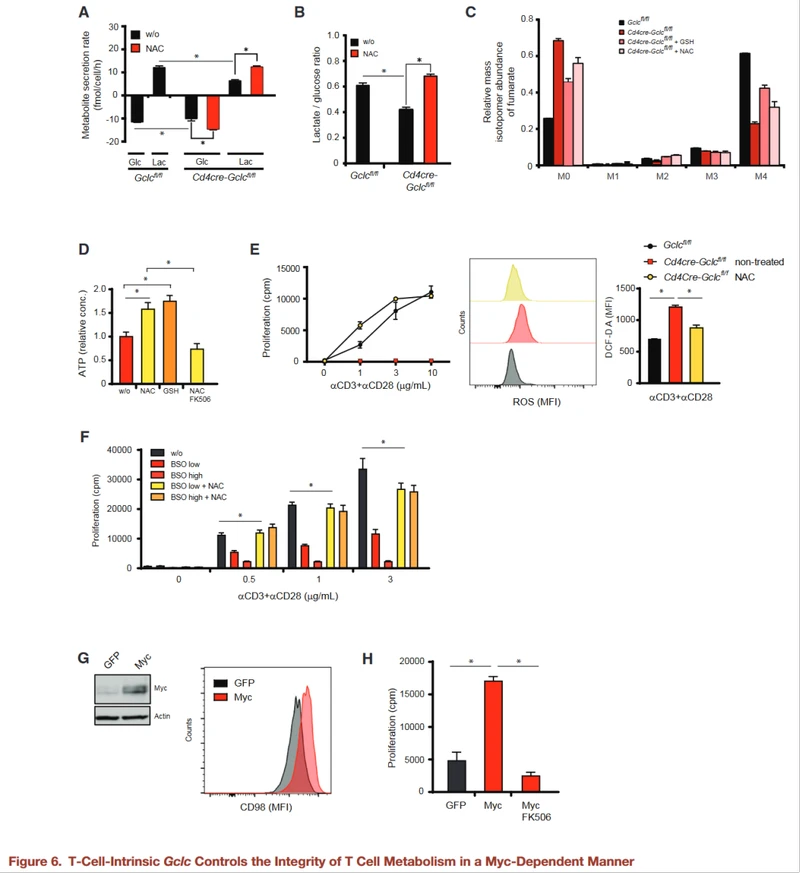
ROS buffering ensures the integrity of T cell energy metabolism
To investigate the effects of antioxidants on T cell metabolism, GCLC-deficient and control T cells were activated in the presence of NAC or glutathione, and glucose consumption and lactate secretion were measured.
Gclc-deficient cells consume less glucose and produce less lactic acid than controls, but their glycolytic activity is reduced in the presence of NAC or glutathione.
These findings were confirmed by U-13C-glutamine measurements of glutamine flux into the TCA cycle. Glucose and glutamine fluxes in activated WT T cells treated with BSO plus NAC or GSH were measured.
As with GCLC-deficient T cells, pharmacological inhibition of GCL in activated WT T cells reduces glucose and glutamine fluxes, while NAC or glutathione restores these fluxes.
Glutathione is essential for ROS buffering and enables metabolic reprogramming during T cell activation.
ROS clearance in activated GCLC-deficient T cells also increases ATP and "recharges" these cells, and FK506-mediated NFAT inhibition inhibits this way of charging.
"Recharged" Gclc deficient T cells treated with antioxidants triggered an activation-induced proliferation response and contained fewer ROS than untreated activated control T cells. WT T cells treated with BSO showed reduced proliferation, which was restored by NAC.
As expected, proliferation of activated GCLCS deficient T cells treated with NAC was inhibited by either FK506 or rapamycin, suggesting that proliferation is NFAT and mTOR dependent in the presence of antioxidants.
To confirm that Myc was indeed affected by Gclc deletion, GCLC-deficient T cells were transduced with MYc-expressing retroviruses for 24 hours.
Compared with controls for GFP transduction, the surface expression of the Myc target CD98 was up-regulated on myC-expressing Gclc deficient T cells. Retroviral Myc expression also restored the proliferation of activated GCLC-deficient T cells.
Since the proliferation of myC-expressing GCLC-deficient cells is dependent on NFAT, there must be a tight interaction between these pathways in T cells.
It is concluded that glutathione maintains T cell metabolic integrity by supporting Myc expression, which is closely related to T cell function.
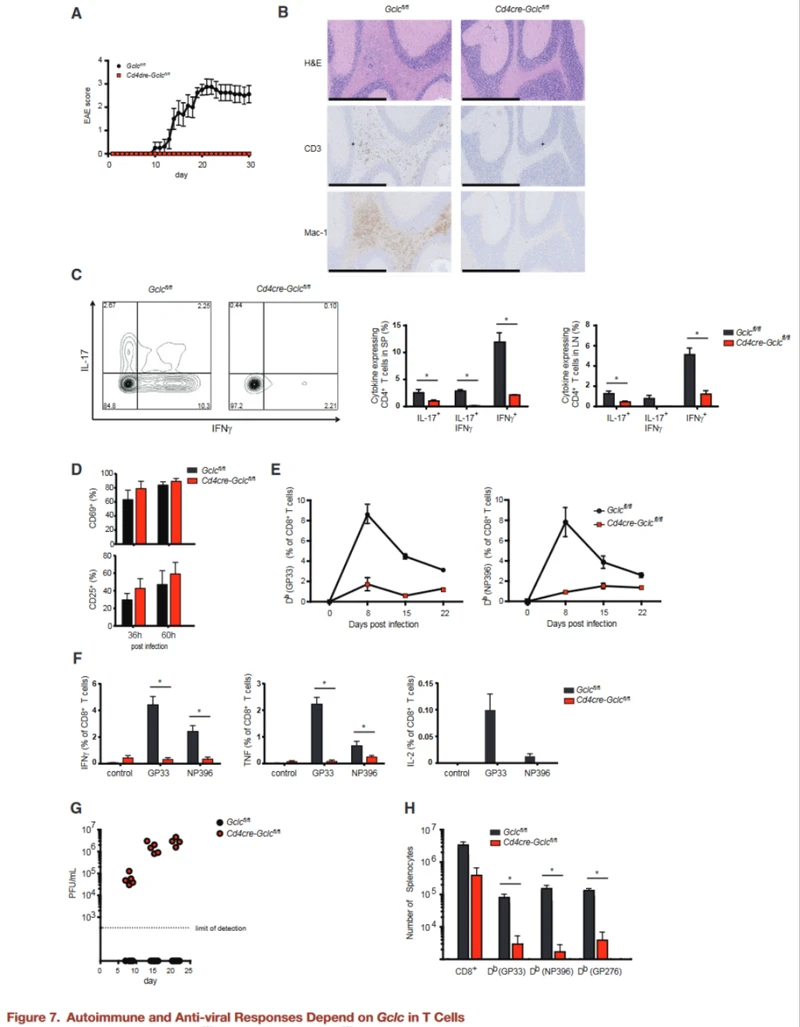
Autoimmune and antiviral responses depend on Gclc in T cells
Next, we explored the consequences of metabolic imbalances in T cells lacking Gclc in vivo.
Because Gclc activity affects T cell homeostasis, which can be severely disturbed in autoimmune diseases, the authors investigated whether targeting the glutathione-dependent pathway might be a promising approach to treating autoimmunity.
Experimental autoimmune encephalomyelitis (EAE) is a mouse model of multiple sclerosis in which CD4+ T cells are the primary driver of inflammation.
We induced EAE in WT and GCLC-deficient mice and immunized them subcutaneously (s.c.) with MOG peptide emulsion (MOG) plus pertussis toxin (PT) to induce disease.
As expected, the Gclcfl/fl mice developed severe EAE on day 30 (d30) after induction, while no GCLC-deficient mice showed any signs of EAE during this period.
Histopathological analysis of brain tissue on day 30 after MOG immunization revealed immune cell infiltration in the control brain and spinal cord, but not in the brain and spinal cord of GCLC-deficient mice.
The infiltrates in the white matter of the control (but not mutated) brain and spinal cord consist primarily of CD3+ T cells and activated macrophages or microglia.
IL-17 and IFN-γ secretion from GCLC-deficient CD4+ T cells was significantly reduced compared to the control group. Therefore, Gclc is essential for stimulating inflammatory T cell responses in an autoimmune setting.
Gclc-deficient mice were unable to develop EAE, suggesting that they may lack a natural T-cell-mediated defense against viral infection.
The authors measured T cell activation in vivo after lymphocytic choriomeningitis virus (LCMV-Armstrong) attack.
CD8+ T cells isolated from LCMV-specific (GP33 antigen) TCR transgenic (P14) Gclcfl/fl or Cd4creGclcfl/fl mice were labeled with CFSE or Purple Cell Trace (VCT), respectively. And transferred an equal number of these cells into the recipient mice.
Splenic T cell activation was measured 36 and 60 hours after these receptors were infected with LCMV. Gclc deficiency does not prevent the initial activation of LCMV-specific T cells in vivo.
When Gclc deficient and control mice were infected with LCMV and the frequency of antigen-specific CD8+ T cells was monitored by tetrimer staining (GP33, NP396) and flow cytometry, significant differences in antiviral response were found.
In control mice, LCMV reactive (GP33+, NP396+) T cells peaked on day 8 after infection, but only a very small number of such T cells were detected in GCLC-deficient mice, and very few of them produced inflammatory cytokines.
The control mice cleared the virus on day 8 after infection, while the virus titer remained high in the mutants. At day 60 after infection, LCMV-specific memory CD8+ T cells (CD44high, CD62Lhigh, CD127high, KLRG1low) and cytokine producing cells were still significantly reduced in GCLC-deficient mice compared to controls.
In summary, activated T cells adjust their metabolism to meet their increased bioenergy and biosynthesis needs. Activated T cells produce ROS, which triggers the antioxidant glutathione response to prevent cell damage, whereas Gclc deficiency results in glutathione reduction, ROS accumulation impinges on mTOR and NFAT activation and downstream cMyc expression, impinges on normal glycolysis and glutamine metabolism, and thus affects inflammatory T cell response.